Entry
Reader's guide
Entries A-Z
Subject index
Chromosomal Microarray
Genetic testing methods have changed over the years. First, there was the advent of chromosome banding methods in the 1970s to 1980s. In the 1990s, there was the isolation of DNA markers from individual chromosomes that were used with fluorescence in situ hybridization (FISH) for identification of deletions too small to be detected in routine chromosome studies. Advances in genetic technology since the 1990s initially led to a collection of thousands of DNA markers from the 23 pairs of human chromosomes for development of chromosomal microarrays. This genetic tool further identified copy number variation or submicroscopic deletions or duplications. Later, the DNA markers included probes to detect single nucleotide polymorphisms (SNPs) as well as chromosome deletions and duplications. Now these high-resolution chromosome microarrays contain over two million probes that can detect very small submicroscopic deletions and duplications as well as DNA polymorphisms, which are useful in research and in clinical studies of common or rare genetic diseases, including many developmental and intellectual disorders. This entry discusses the use of chromosome microarrays.
Use of Chromosome Microarrays
High-resolution microarrays are now in common use in identifying genetic defects and patterns in individuals presenting for clinical services with intellectual disability and/or autism spectrum disorders (ASD). Thus, the rise of chromosomal microarrays or array comparative genomic hybridization (aCGH) with copy number probes and later high-resolution microarrays with both copy number and SNP probes have significantly impacted understandings of genetic factors contributing to human disease.
For example, studies have shown that as high as 90% of those with ASD may have causative genetic factors. Furthermore, probes for SNPs, which can detect differences in the DNA pattern from individual to individual, are helpful in identifying regions of homozygosity or areas of DNA that appear identical. These stretches of DNA contain genes that are identical by descent or coming from a common ancestor, thereby supporting consanguinity or inbreeding, particularly when these regions share loss of heterozygosity not due to missing or deleted DNA segments on the other member of the chromosome pair. An example of loss of heterozygosity and not due to inbreeding or a deletion is a genetic phenomenon referred to as uniparental disomy when both members of a chromosome pair come from only one parent as seen in Prader-Willi syndrome (PWS), a classical but rare genetic disorder with maternal disomy 15 being the second most common cause of the disorder. A chromosome 15q11-q13 deletion involving the father’s chromosome 15 is the most common cause (see Figure 1).
Figure 1 Chromosome Microarray Results for Prader-Willi Syndrome
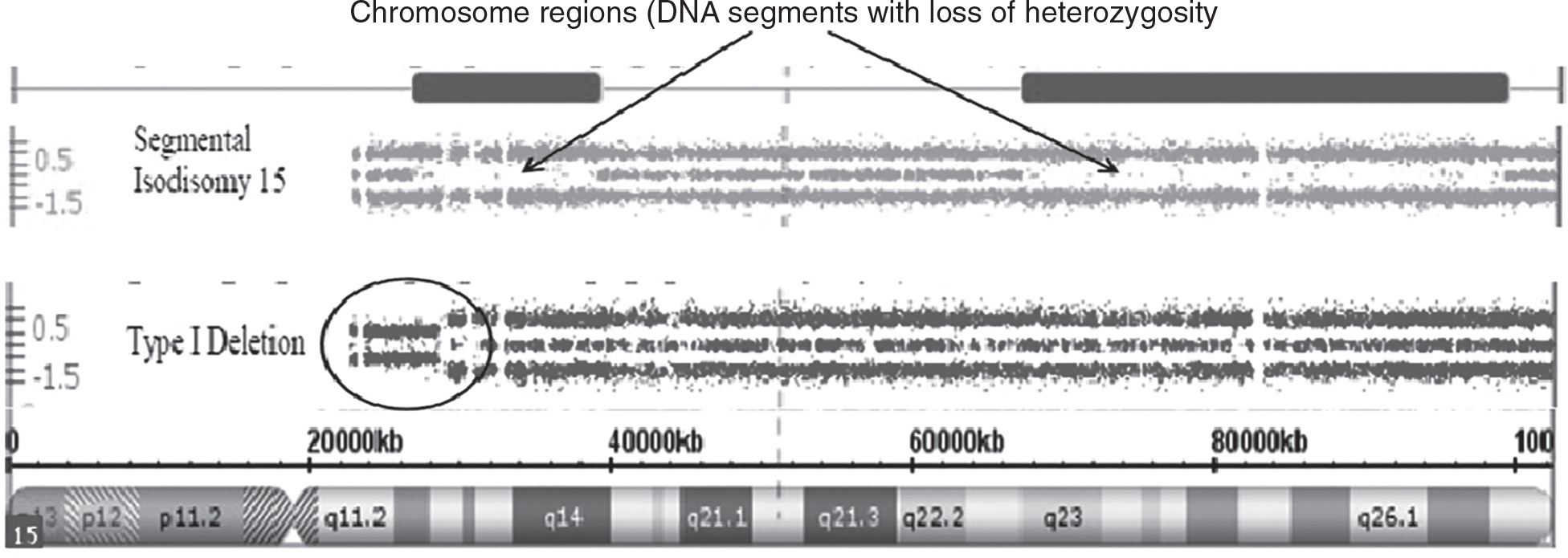
Source: Merlin G. Butler and A. M. Manzardo
Consanguinity is a common occurrence in some cultures, and the offspring from consanguineous or related parents share common gene alleles and are at risk of inheriting genes with defects or mutations that can cause genetic diseases, particularly autosomal recessive conditions such as cystic fibrosis. Consanguinity is termed as a union between individuals who are biologically related at the level of second cousins or closer. The degree of consanguinity can be based on the amount of homozygosity or lack of DNA polymorphic signals in the genome and assessed by using SNP data from high-resolution microarrays to calculate the coefficient of inbreeding. Couples related as second cousins or closer and their progeny account for an estimated 10% of the global population.
...
- Assessment and Diagnosis
- Diagnostic and Statistical Manual of Mental Disorders
- International Classification of Diseases, 10th Revision
- International Classification of Diseases, History of
- Academic Achievement Tests
- Adaptive Behavior Assessment System
- Adults With Autism Spectrum Disorder
- Assessment of the Very Young Child
- Attention-Deficit/Hyperactivity Disorder: Assessment of Children With
- Autism Diagnostic Interview-Revised
- Autism Diagnostic Observation Schedule
- Autism Rating Scales
- Autism Spectrum Disorder: Assessment in Schools
- Autism Spectrum Disorder: Assessment of Adults With
- Autism Spectrum Disorder: Assessment of Comorbid Psychiatric Conditions
- Autism Spectrum Disorder: Assessment of Social Behavior
- Autism Spectrum Disorder: Assessment of Speech, Language, and Communication Disorders in
- Autism Spectrum Disorder: School Consultation
- Autism Spectrum Rating Scales
- Bayley Scales of Infant and Toddler Development
- Behavior Checklists
- Child Behavior Checklist
- Chromosomal Microarray
- Cognitive Assessment System
- Comprehensive Executive Functioning Inventory
- Continuous Performance Tests
- Developmental Interview
- Diagnostic Reading Assessment
- Differential Ability Scales
- Dyslexia: Assessment
- Executive Function Tests
- Expressive Language Tests
- Eye Tracking
- Floortime
- Functional Behavioral Analysis
- Genetic Counseling
- Giftedness
- Infant Visual Attention and Response to Novelty
- Intellectual Disability, Assessment of Adults With
- Intellectual Disability, Assessment of Children With
- Intelligence-Achievement Discrepancy
- Intelligence: IQ Scores
- Labeling, Pros and Cons of
- Magnetic Resonance Imaging
- Mental Status Examination
- Mullen Scales of Early Learning
- Neuroimaging Methods
- Neurological Examination
- Neuropsychological Assessment
- Newborn Screening
- Noninvasive Brain Stimulation
- Nonverbal Assessment of Intelligence
- Processing Speed
- Psychological Testing
- Receptive Language Tests
- Stanford-Binet Intelligence Scales
- Structured Interviews
- Testing for Prenatal Birth Defects
- Vineland Adaptive Behavior Scales
- Wechsler Adult Intelligence Scale
- Wechsler Individual Achievement Test
- Wechsler Intelligence Scale for Children
- Wechsler Preschool and Primary Scale of Intelligence
- Whole Genome Sequencing
- Woodcock-Johnson Tests of Achievement
- Woodcock-Johnson Tests of Cognitive Abilities
- Written Expression, Tests of
- Comorbidity and Associated Features
- Aggression
- Alcohol Abuse in Individuals With Intellectual and Developmental Disabilities
- Allergies
- Aphasia
- Apraxia
- Blindness
- Cleft Lip and Cleft Palate
- Deafness and Hearing Loss
- Depressogenic Conditions
- Diagnostic Overshadowing
- Dual Diagnosis
- Echolalia
- Empathy
- Encopresis
- Enuresis
- Executive Functioning
- Fine Motor Delays
- Gross Motor Delay
- Hyperkinesis
- Hypertonia and Hypotonia
- Infantile Automatisms
- Joint Attention
- Mania
- Munchausen by Proxy
- Nonepileptic Stress Induced Seizures
- Obesity
- Obsessions and Compulsions
- Otitis Media
- Panic Attacks
- Peer Rejection
- Perseveration
- Phobias
- Pica
- Pragmatics
- Psychosis
- Regression
- Repetitive Behaviors and Interests
- Resilience, Learning Disorders and
- Seizures
- Self-Injury
- Self-Stimulatory Behaviors
- Sensory Integration Issues
- Sexual Abuse
- Sexual Exploitation
- Sexualized Behavior
- Siblings of Individuals With Developmental Disorders
- Sleep Disorders
- Social Anxiety
- Social Skills Deficits
- Stereotypy
- Stuttering
- Substance Use
- Suicide
- Trauma
- Development
- Adolescence
- Cognitive Development
- Developmental Milestones
- Early Childhood Development
- Elementary School-Aged Children With Intellectual and Developmental Disabilities
- Emotional Development
- Fine Motor Development
- Gross Motor Development
- Infancy
- Intelligence: Environmental Factors
- Memory
- Metacognition
- Moral Development
- Nature and Nurture
- Neurobiology of Developmental Disabilities
- Phenotype
- Piaget’s Theory of Cognitive Development
- Preschool Intellectual and Developmental Disorders
- Readiness Skills
- Self-Regulation
- Social Cognition
- Social Skills
- Speech and Language Development
- Temperament
- Theory of Mind
- Transitional Age Youth
- Visual Motor Coordination
- Visual Spatial Skills
- Developmental and Intellectual Disorders
- 16p11.2 Deletion and Duplication Syndromes
- 18p Deletion Syndrome
- 22q11.2 Deletion Syndrome
- Acute Disseminated Encephalomyelitis
- Adrenoleukodystrophy and Other Leukodystrophies
- Agenesis of the Corpus Callosum
- Alice in Wonderland Syndrome
- Asperger Syndrome
- Autism Spectrum Disorder
- Cerebral Palsy
- Childhood Disintegrative Disorder
- Chromosome 15 Disorders
- Cornelia de Lange Syndrome
- Cri-du-Chat Syndrome
- Dandy-Walker Syndrome
- Down Syndrome
- Epilepsy
- Failure to Thrive
- Fetal Alcohol Spectrum Disorders
- Fragile X Syndrome
- Global Developmental Delay
- Intellectual Disability
- Intellectual Disability, Abuse and
- Intellectual Disability: Comorbidity
- Intellectual Disability: Development and Outcome
- Intellectual Disability: Mild
- Intellectual Disability: Moderate
- Intellectual Disability: Severe and Profound
- Klinefelter Syndrome
- Landau-Kleffner Syndrome
- Learning Disorders: Relationship to Other Neurodevelopmental Disorders
- Mitochondrial Disorders
- Multiple Sclerosis
- Muscular Dystrophies
- Neonatal Abstinence Syndrome
- Neurofibromatosis
- Noonan Syndrome
- Patau Syndrome
- Pitt-Hopkins Syndrome
- Prader–Willi Syndrome
- Rett Syndrome
- Rubinstein-Taybi Syndrome
- Shaken Baby Syndrome
- Smith-Magenis Syndrome
- Spina Bifida
- Stroke, Prenatal
- Strokes, Perinatal and Pediatric
- Sturge-Weber Syndrome
- Subacute Sclerosing Panencephalitis
- Triple X Syndrome
- Trisomy
- Tuberous Sclerosis
- Turner Syndrome
- Williams Syndrome
- Wilson’s Disease
- Education
- 504 Plan
- Accommodations
- Adult Literacy
- American Sign Language
- Autism Spectrum Disorder: School Consultation
- Camp Shriver
- Case Management and Care Coordination
- Early Childhood Education for Students With Developmental Disabilities
- Early Intervention
- Early Start Denver Model
- Educational Advocacy
- Elementary Education for Students With Developmental Disabilities
- Employment for Individuals With Autism Spectrum Disorder
- Employment for Individuals With Intellectual Disabilities
- Families as Partners in Educational Decision Making
- Free Appropriate Public Education
- Future Research Directions in Developmental Disabilities
- Head Start Programs
- Homeschooling
- Homework
- Inclusion
- Individualized Education Program
- Individualized Family Service Plan
- Individuals with Disabilities Education Act
- Infant Stimulation Programs
- Intelligence: IQ Scores
- Job Coaching
- Learning Disorders: Relationship to Other Neurodevelopmental Disorders
- Least Restrictive Environment, Current Practices in
- Life Skills Coaching
- Mainstreaming, Current Practices in
- Mentoring
- Multidisciplinary Approaches to Teaching Students With Developmental Disabilities
- Online Learning
- Peer Tutoring
- Physical Activity and Children and Youth With Intellectual and Developmental Disabilities
- Portage Project
- Positive Behavior Support and Developmental Disabilities
- Postsecondary Education for Students With Disabilities
- Program Evaluation
- Readiness Skills
- Reading Programs: Teaching Students With Intellectual Disabilities and Moderate-to-Severe Reading Disabilities
- Regular Education Initiative
- Secondary Education for Students With Developmental Disabilities
- Section 504 and the Education of Children With Disabilities
- Self-Contained Classrooms
- Social Skills Groups for Individuals With Autism Spectrum Disorders
- Special Education
- Support Groups for Adults With Intellectual and Developmental Disabilities
- Support Groups for Families of Children With Developmental Disorders
- Supportive Employment
- Technology for Individuals With Intellectual and Developmental Disabilities
- Technology-Enhanced Learning
- Universal Design for Learning
- Gender, Culture, and Ethnicity
- Larry P. v. Riles
- Bilingualism
- Cultural Differences in Discipline
- Cultural Norms
- Culturally Sensitive Assessment
- Famous People With Intellectual and Developmental Disabilities
- Gender Differences
- Intelligence Tests, Effect of Culture and Ethnicity on
- International Perspectives on Developmental Disabilities
- Maltreatment of Persons With Intellectual Disabilities
- Media Portrayals of Individuals With Developmental Disabilities
- Parenting Style
- People-First Language
- Race and Ethnicity
- Stigma
- Student–Teacher Relationships
- Health, Wellness, and Resiliency
- Activities of Daily Living
- Allergies
- Bullying
- Commercial Sexual Exploitation of Children
- Dating and Close Relationships
- Emotional Abuse
- Employment for Individuals With Autism Spectrum Disorder
- Employment for Individuals With Intellectual Disabilities
- End-of-Life Care
- Estate Planning
- Family Impact in Adulthood
- Friendships
- Funding Services for Adults With Developmental Disabilities and Autism Spectrum Disorder
- Giftedness
- Health Promotion
- Health-Related Disorders
- Identity Development
- Intellectual Disability: Prevention
- Learned Helplessness
- Life Skills
- Lifespan Developmental Theories
- Long-Term Supports and Services
- Parental Adaptation
- Parental Stress
- Partnering With Refugees With Intellectual and Developmental Disabilities and Their Families
- Peer Acceptance
- Physical Abuse
- Physical Activity and Children and Youth With Intellectual and Developmental Disabilities
- Prevention of Intellectual and Developmental Disabilities
- Protective Factors
- Quality of Life
- Resilience, Characteristics of
- Respite Care
- Self-Esteem
- Sexual Awareness and Knowledge
- Sexual Development, Physical
- Sexuality in Autism Spectrum Disorder
- Sexuality in Children and Adolescents With Developmental Disorders
- Sexuality in Intellectual Disabilities
- Social Support
- Special Olympics
- Splinter Skills and Cognitive Strengths in Autism
- Sports
- Stress and Coping
- Supported Living
- TEACCH
- Transitional Age Youth
- Vaccinations and Autism
- Historical Views of Intelligence and Development
- Diagnostic and Statistical Manual of Mental Disorders
- International Classification of Diseases, 10th Revision
- International Classification of Diseases, History of
- Autism and Autism Spectrum Disorders, History of
- Civil Rights of Institutionalized Persons Act
- Crystallized Intelligence
- Deinstitutionalization
- Disability Rights Movement
- Education for All Handicapped Children Act
- Epidemiology, Historical Changes in
- Eugenics Movement
- Fluid Intelligence
- Flynn Effect
- Head Start, History of
- Intellectual Disability, History of
- Intelligence, Historical Views of
- Least Restrictive Environment, History of
- Mainstreaming, History of
- Special Education, History of
- Learning, Psychological, and Behavioral Disorders
- Adjustment Disorders
- Attention-Deficit/Hyperactivity Disorder
- Bipolar Disorder
- Conduct Disorder
- Depression
- Developmental Coordination Disorder
- Developmental Dyscalculia
- Disorders of Elimination
- Disruptive Behavior Disorders
- Disruptive Mood Dysregulation Disorder
- Dysgraphia
- Dyslexia
- Dyslexia: Treatment
- Eating Disorders
- Feeding Disorders of Infancy or Childhood
- Generalized Anxiety Disorder
- Language Disorders
- Learning Disorders
- Nonverbal Learning Disability
- Obsessive-Compulsive Disorder
- Oppositional Defiant Disorder
- Panic Attacks
- Panic Disorder
- Pediatric Autoimmune Neuropsychiatric Disorders Associated With Streptococcal Infections
- Posttraumatic and Acute Stress Disorders
- Rumination Syndrome
- Schizophrenia, Adult Onset
- Schizophrenia, Early Onset
- School Refusal
- Selective Mutism
- Separation Anxiety Disorder
- Social (Pragmatic) Communication Disorder
- Social Anxiety
- Specific Phobia
- Speech Sound Disorder
- Tic Disorders
- Tourette’s Disorder
- Public Policy Issues
- Larry P. v. Riles
- Americans with Disabilities Act
- Arc of the United States, The
- Article 12 of the Convention on the Rights of Persons with Disabilities
- Camp Shriver
- Classroom Aides
- Conservatorship
- Council for Exceptional Children
- Death Penalty and Intellectual Disability
- Decision-Making Capacity
- Dispute Resolution
- Down Syndrome: Cognitive, Emotional, and Behavioral Presentation
- Down Syndrome: Medical Concerns and Comorbid Disorders
- Education for All Handicapped Children Act
- Ethical Issues in Education
- Ethical Issues in Research, Current Standards in
- Ethical Issues in Research, History of
- Ethical Issues in Treatment
- Federal Funding for Children With Intellectual and Developmental Disabilities
- Financial Abuse
- Free Appropriate Public Education
- Funding Services for Adults With Developmental Disabilities and Autism Spectrum Disorder
- Guardianship
- Inclusion
- Labeling, Impact of
- Labeling, Pros and Cons of
- Section 504 and the Education of Children With Disabilities
- Special Education Law: U.S. Supreme Court Cases
- Research
- Consent in Research and Treatment
- Disability Research Methodology
- Epidemiology
- Familial Aggregation Studies
- Flynn Effect
- Future Research Directions in Developmental Disabilities
- Genetic Research
- Longitudinal Research
- Multiple Baseline Design
- Naturalistic Observation
- Randomized Controlled Trials
- Single Case Research Design
- Treatment Intervention
- Theories and Causes: Biological Perspectives
- Acquired Pediatric Brain Damage
- Anoxia
- Autism Spectrum Disorder, Biology of
- Basal Ganglia
- Birth Defects
- Brain Plasticity
- Brain Tumors, Pediatric
- Brainstem
- Central Nervous System
- Cerebellum
- Cerebral Cortex
- Cingulate Cortex
- Cognitive Neuroscience of Intelligence
- Concussion
- Corpus Callosum
- Cross Disorder Genetic Risk
- Developmental Psychopathology
- Down Syndrome: Medical Concerns and Comorbid Disorders
- Encephalitis
- Endophenotypes
- Failure to Thrive
- Frontal Lobes
- Genetics of Intellectual Development
- Hemispheric Asymmetries
- Herpes Simplex
- Hormones
- Human Genome Project
- Hydrocephalus
- Iatrogenic Effects
- Intellectual Disability, Biology of
- Lead Poisoning
- Learning Disorders, Biology of
- Limbic System
- Maternal Substance Abuse
- Media Use by Individuals With Intellectual and Developmental Disabilities
- Meningitis
- Microcephaly
- Monogenic Disorders
- Mosaicism
- Myelination and Connectivity
- Neural Plasticity
- Neuroimaging Methods
- Neurotransmitters
- Nutrition in Childhood and Adolescence
- Nutrition, Prenatal
- Nutrition: Importance in First Years of Life
- Polygenic Risk
- Postnatal Risk Factors
- Prenatal Risk Factors
- Preterm Birth
- Rubella
- Temporal Lobe
- Teratogens
- Theories and Causes: Psychological and Sociocultural Perspectives
- Attachment Theory
- Autism, Rising Prevalence of: Epidemiology
- Autism, Rising Prevalence of: Theories
- Disability Discrimination: U.S. Supreme Court Cases
- Down Syndrome: Cognitive, Emotional, and Behavioral Presentation
- Ecological Systems Theory
- Environmental Considerations in Social Participation of Children With Developmental Disabilities
- Environmental Risk Factors
- Erikson’s Theory of Psychosocial Development
- Gardner’s Theory of Multiple Intelligences
- Intelligence, Theories of
- Intelligence: Environmental Factors
- Piaget’s Theory of Cognitive Development
- Vulnerability Stress Model
- Vygotsky’s Zone of Proximal Development
- Therapies and Interventions
- Access to Treatment for Individuals With Intellectual and Developmental Disabilities
- Accessible Design
- Adaptive Functioning
- Anchored Instruction
- Applied Behavior Analysis
- Art Therapy
- Assistive Technology
- Attention-Deficit/Hyperactivity Disorder: Potential Misuse of Stimulant Medications
- Augmentative and Alternative Communication
- Behavior Modification
- Behavior Therapy
- Biofeedback
- Braille, Use in Individuals With Visual Impairment
- Classroom Aides
- Cognitive Behavioral Therapy
- Cognitive Rehabilitation Training
- Community-Based Interventions
- Consent in Research and Treatment
- Dementia
- Dementia and Aging in Down Syndrome
- Discrete Trial Training
- Early Intervention
- Early Start Denver Model
- Executive Function Coaching
- Extinction
- Family-Based Interventions
- Flooding
- Floortime
- Functional Inclusion
- Intellectual Disability: Historical Changes in Terminology
- Male Brain Theory of Autism
- Mentoring
- Metacognitive Training
- Mindfulness-Based Treatments
- Mood Stabilizers
- Music Therapy
- Nutritional Therapies for Children With Developmental Disabilities
- Occupational Therapy
- Parent–Child Interaction Therapy
- Personalized Supports
- Phonics-Based Interventions
- Physical Therapy
- Picture Exchange Communication System
- Pivotal Response Therapy
- Portage Project
- Positive Parenting of Children With Intellectual and Developmental Disabilities
- Prenatal Care
- Probiotics
- Psychopharmacological Interventions
- Relationship Development Intervention
- Social Inclusion
- Speech and Language Pathologists
- Stimulant Medications
- Systematic Desensitization
- Technology for Individuals With Intellectual and Developmental Disabilities
- Treatment Planning
- UCLA Young Autism Project
- Loading...
Get a 30 day FREE TRIAL
-
 Watch videos from a variety of sources bringing classroom topics to life
Watch videos from a variety of sources bringing classroom topics to life -
Read modern, diverse business cases
-
Explore hundreds of books and reference titles

Read next

More like this
Sage Recommends
We found other relevant content for you on other Sage platforms.
Have you created a personal profile? Login or create a profile so that you can save clips, playlists and searches